●計算例1 PM3計算による構造最適化と生成熱・双極子モーメントの計算
- MOPACメニューからMinimize Energyを選ぶ。
- RMSは0.100のまま使用し,TheoryのMethodでハミルトニアンとしてPM3を選択する(図1)。
- ProperitiesリストでDipole(双極子モーメント)とHeat of Formation(生成熱;MOPACでは,298K標準状態における気体相の値)を選択する(図2)。複数の因子を選ぶ場合はCTRLキーを押しながらクリックする。
- Runボタンをクリックして計算を実行する。計算終了後は分子を任意の形式で保存する。MDL MOL形式で保存すればChimeでの利用が可能である。
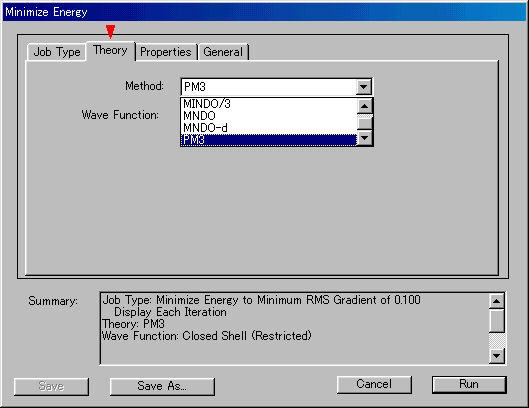 図1
図1
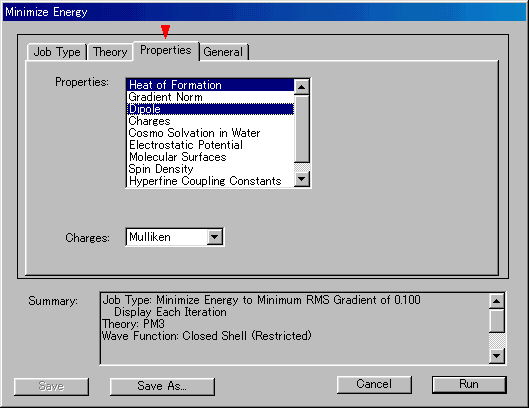 図2
図2【計算結果例】
- 水(H2O)の双極子モーメント
- PM3: 1.739 debye
- AM1: 1.861 debye
- ホルムアルデヒド(HCHO)の双極子モーメント
- PM3: 2.164 debye
- AM1: 2.317 debye
- 2-ナフトール(β-ナフトール)の生成熱と双極子モーメント
分子モデル (a) 空間充填 球棒 スティック 針金
背景・黒 灰 白 紺
(b) 空間充填 球棒 スティック 針金
背景・黒 灰 白 紺生成熱 / kcal mol-1 PM3 -4.36065 -4.68730 AM1 -3.47033 -3.93693 双極子モーメント / debye PM3 1.369 0.872 AM1 1.460 0.987
※他サイトの参考Chime分子
- aタイプまたはaに近いタイプの2-ナフトール
- 他の形をとる2-ナフトール
●計算例2 LUMO・HOMOエネルギー計算と軌道の可視化
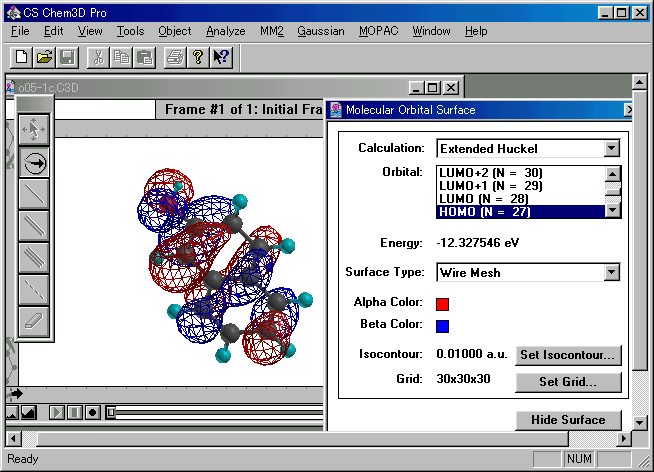
図3 2-ナフトールのHOMOの例
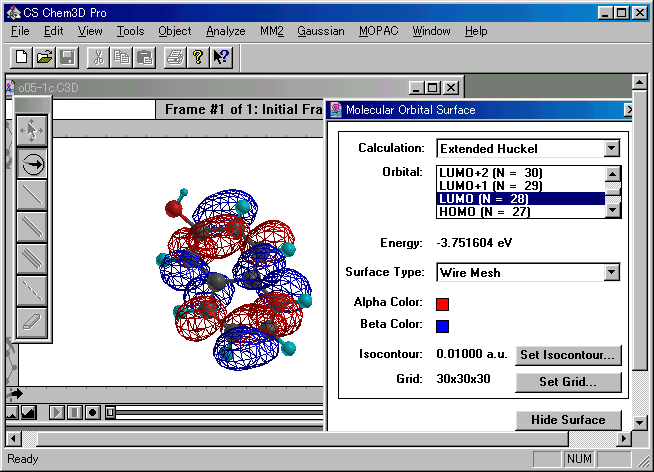
図4 2-ナフトールのLUMOの例
フロンティア軌道理論では,ある分子内で求電子反応,求核反応,ラジカル反応が起こる位置を,次のように予想します.
●計算例3 WinMOPAC/MOS-Fによる励起エネルギー計算
◆杉野圭司,「パソコン分子設計 応用編」,p.73,サイエンスハウス(2000) より
(a) 求核的攻撃を受けやすいのは,基底状態のLUMOの電子密度が最大の位置.
(b) 求電子的攻撃を受けやすいのは,基底状態のHOMOの電子密度が最大の位置.
(a) ラジカル的攻撃を受けやすいのは,HOMOとLUMOに電子が1個ずつ入ったときの電子密度の和(すなわち,SOMOの電子密度)が最大の位置.
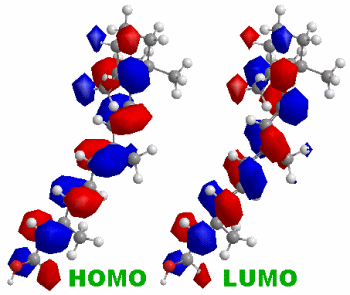
※参考:Chem3D Version 10.0(2006年購入)によるall-trans-ビタミンAのHOMOとLUMOの表示
 演習例メニュー
演習例メニュー  分子の組み立て
分子の組み立て  可視吸収スペクトルとその計算
可視吸収スペクトルとその計算